

Ми зупинились на тому, що геноми складені до купи, там знайдені відповідні гени та, можливо, інші фічі, геноми складаються в бази даних. Як це все тепер виглядає для простогокористувача?
Як правило, під кожний референтний геном готується так званий геномний браузер, який я вже згадувала раніше.

Це ніщо інше, як графічна візуалізація бази геномних даних. Вони різняться по дизайну, але у більшості з них одні і ті самі принципи. Можна просто зумити певну ділянку хромосоми. Гени часто перелінковані на інши бази даних, де можна знайти інформацю про роботу цього гену, а також про відомі мутації і, якщо пощастить, про фенотипові прояви цих мутацій. З цієї бази даних можна легко і просто отримати задану послідовність ДНК - в окремому вікні є опція - dump FASTA file, після чого з означеної ділянки ДНК формується придатний до скачування текст геному. Також в пошуковому вікні браузера можна завдавати пошук координат (конкретна хромосома і відстань в нуклеотидах), пошук по простій назві гену або по оригінальному identifier (кожен ген, як правило, має унікальний номер).
Наприклад, identifier гену арабідопсису виглядає так: At3G01090. At означає Arabidopsis thaliana, далі йде номер хромосоми, а потім п'ятизначне число, яке вказує на орієнтацію на хромосомі. У різних організмів різні номенклатурні принципи, тому збираються консорціуми, аби призвести це до спільного знаменника.
Ось тут декілька прикладів геномних бразуерів - можете переконатись, що кроєні вони по одному лекалу.
Дрозофіла, курка, жабка, щур, кукурудза, людина.
Як вже зрозуміло, для маглів девайс зовсім непридатний. Я собі уявляю в майбутньому щось легке і зрозуміле, на кшалт фільтру по клінічно релевантній інформації.
Гаразд, а що роблять з прочитаним геномом науковці? Перш за все відбувається просте описання і класифікація тих генів, які там познаходили: їх кількість, класи, сімейства. Уважно розглядають, де вони розташовані.
Наприклад, ось так виглядає самий перший зум. Це схематичне зображення геному картоплі. Зовнішнє коло діаграми - це фото хромосом, де червоною рисочкою позначені центромери (це місце, де

Наступне врутрішнє коло - кольорова схема щільності генів. Як ми бачим, поблизу центромери світлозелений колір, щільність генів низька (це взагалі відомий факт), при цьому чим далі від центромери до хвостів, тим щільність зростає. А от кількість повторів (третє ззовні коло) - навпаки. Найбільше повторів саме біля центромер.
Після того, як це все описали і класифікували, добре було б мати ще декілька інших геномів для порівняння. З цього моменту до описової геноміки додається порівняльна, або компаративна. На компаративній геноміці базується міжвидове або внутрішньовидове еволюційне порівняння геномів, а також популяційні дослідження про розповсюдження мутацій або інших геномних змін.
Цумбайшпіль, порівняли геноми арабідопсису, картоплі і винограду.

Дивимось на малюнок С. Що ми бачим? Ми бачим, що геноми цих трьох рослин місцями перекриваються. Таким чином, порівнюючи віддалені геноми, можна знаходити гени, які дуже схожі в усіх рослин. Вони називаються древніми, консервативними і вважається, що вони відіграють ключову роль у живих організмів. В той же час, дехто навпаки, шукає специфічні гени для того, чи іншого організму - це саме ті гени, які зумовили оце різноманіття.
Еволюційне порівняння окремих генів роблять за допомогою Clustal алгоритму або методом приєднання сусідів.
Це все, звісно, дуже приблизно, бо в природі трапляється різне - гени після маленьких мутацій можуть відігравати зовсім іншу роль в новому організмі. Або навпаки - глобальні геномні перебудови не мають великих наслідків.
Або ось ще один приклад. Порівняли геноми різних сортів картоплі: кругленьку і довгасту.

На малюнку c візуалізація порівнянь по хромосомах, на малюнку d - зум так, щоб можна було уважніше роздивитись конкретні геномні перебудови на прикладі хромосоми 5. Видно, що деякі гени подвоїлись (зліва на малюнку синя стрілочка С1 і С2), деякі змутували (червоні стрілочки), деякі - присутні тільки в одному з сортів (зелені).
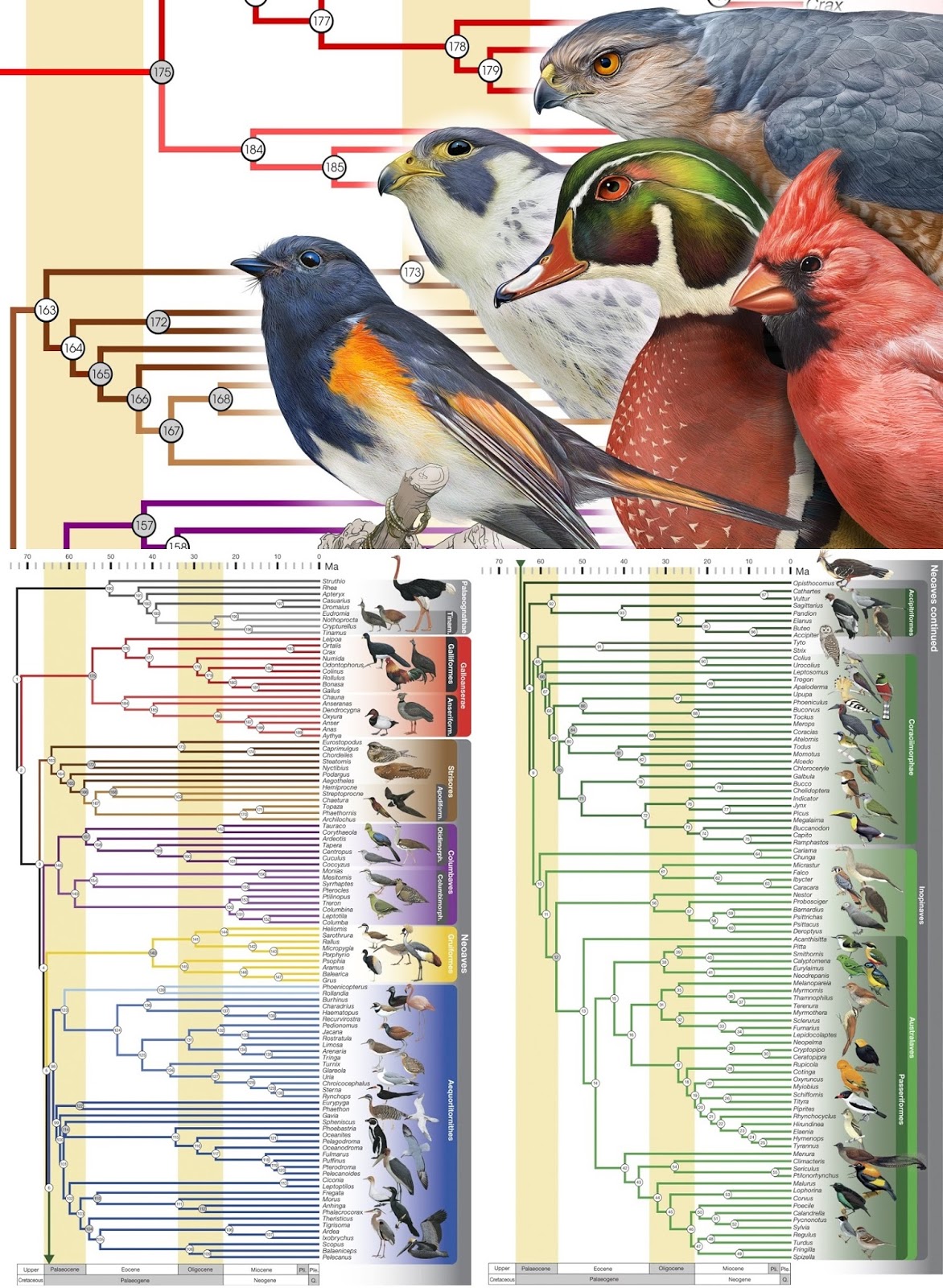
Що стосується компаративної і еволюційної геноміки, ще один свіжий приклад. Поки я писала цей пост, в фб кинули лінк на філогенію 198 видів пташок, яка базується на секвенуванні їх геномів. Взагалі цікава стаття, бо в методах дає повний огляд пайплайну, про який ми тут говорили. Спочатку взяли геном курки (з лінку вище) і вибрали з нього консервативний геномний локус, по якому проводили порівняння. Потім виділили з пташок ДНК, збагатили його на цей локус і прочитали його секвенуванням. Далі робили ассемблінг, причому як по референтному геному, так і de novo. Філогенетичне порівняння робили вищезгаданим neighbor joining в різних варіаціях.
На цьому завершимо порівняння пташок і перейдем до третього важливого вивчення геному - популяційної варіації.

Якщо взяти геноми двох людей, то вони будуть трішки різні. Крім великих вставок або делецій, приблизно кожний 1000й нуклеотид буде відрізнятись. Такі одиночні нукледотидні заміни називають "сніпи" від SNPs (single nucleotide polymorphism). Частіше всього вони розташовані в некодуючих ділянках геному, тобто там, де генів мало, або в інтронах або в інших ділянках. Вони можуть бути також розташовані в кодуючій ділянці, викликаючи зміну білку при розшифровці (тоді це буде справжня мутація), або не викликаючи жодних ефектів, якщо цей нуктеотид стоїть, наприклад, третім в кодоні - тоді заміна називається синонімічною.
У кукурудзи, наприклад, такі сніпи зустрічаються значно частіше, фактично кожні 100 нуклеотидів, тому на генетичному рівні різниця між двома сортами кукурудзи більша, ніж між людиною і шимпанзе. Навіщо нам це знати?
Виявилось, що ці сніпи можуть слугувати сигнальними прапорцями - маркерами, які свідчать про якісь інші зміни в геномі, які проявляються в фенотипі, або, як вже я сказала, можуть самі бути мутаціями. Детектувати ці сніпи простіше, ніж секвенувати весь геном.
Якщо взяти дві групи людей, одна з яких, наприклад хвора і дослідити їх сніпи, то може виявитись, що у хворої групи людей частіше зустрічається якийсь конкретний сніп. Це робиться за допомогою наразі дуже модного підходу "джівас" genome wide association study (GWAS). Тоді його називають маркерним і вважають гідним для популяційного скринінгу. Беруть здорову людину, знаходять цей сніп і кажуть, що є ймовірність захворіти. Це мега спрощений дизайн експерименту, бо насправді тут є купа підводних каменів.

Я колись давно писала про це все на пальцях, хоч і трохи в іншому контексті: один, два, три.
Справа в тому, що у людській популяції ми фіксуємо її поточний стан без врахування педігрі. Популяція це суміш поколінь. До інформації про сніп нам потрібно крім фенотипу (хвороби), також історію його спадкування, яку ми накладаємо заднім числом, враховуючи приблизно частоту рекомбінацій, успадкування від батьків, розмір популяції та інші моменти. Там чином рахується linkage disequilibrium і ймовірність (!) асоціації певного сніпу до фенотипу.
Хоч всіх сніпів у людей познаходили близько 150 млн, клінічно релевантних десь 10000, при цьому трешхолд проходить по збільшенню ймовірності захворіти у 2 рази в порівнянню з середньою температурою по палаті. Я так гадаю, що "залізобетонних" сніпів, які гарантують захворювання, значно менше. По факту можна так сказати, що гора родила мишку, успіхи доволі скромні.
Як би там не було, всі релевантні сніпи каталогізовані і їх можна роздивитись в базі даних. Власне ця штука і є першим підходом персоналізованої медицини.

Однак попереду у нас, як ми пам'ятаємо, гігантські плани прочитки людських геномів. Можливо там буде більш обнадійлива ситуція з дизайном експериментів. Наразі там чорт ногу зломить, поки що розмов про потенціал бігдата і персоналізовану медицину більше, ніж вихлопу. Точаться дискусії про те, як зупинити лавину наукоподібного сміття.
У рослин з цим справа дещо краща. У них більше сніпів, їх можна гарненько дослідити в поколіннях, схрещуючи контрастні фенотипи, а потім додатково схрещувати дітей і батьків, відслідковуючи сніп і фенотип стійкості до довгоносика. Ці сніпи використовують в marker assisted selection, коли генетичний аналіз по маркерам, сніпам в тому числі, дозволяє відібрати рослини з певною ознакою задовго до того, коли вона проявиться. По лінку вище демонстрація - як проходить сучасний селекційний процес. Тема модна, перспективна і на неї кинуто купу фінансових і інтелектуальних зусиль.
На цій оптимістичній ноті завершимо про геноміку і перейдем до транскриптоміки.
Для початку освіжим шкільний матеріал. З довгої молекули ДНК зчитується коротенька РНК, яка складається з також з нуклеотидів з однією різницею, що замість тіміну вбудовується урацил (це називається транскрипція), а з неї білок, що складається з амінокислот (трансляція).

З однієї ділянки ДНК зчитується велика кількість молекул матричної РНК. Ми можем ці молекули виділити і порахувати різними методами. Якщо ми спостерігаєм, що в певний момент часу в клітині є матрична РНК з якогось гену, то вважаємо, що ген "включився" і працює. Чим більше молекул, тим сильніше працює ген. Це називається рівнем експресії гену.
Найновіші аналітичні методи дозволяють спостерігати роботу фактично усіх генів одночасно в певний момент часу. Найкраще спостерігати цю роботу в порівнянні: два сорти рослин, дві тканини з різних частин тіла, або одна та сама тканина у здорової людини або пацієнта.
Метод перший - біочіпи. На тверді носії, приміром предметне скло, робот наносить мініатюрні цяточки краплини, які містять насинтезовані штучно коротенькі ділянки ДНК, що відповідають певному гену (фото зліва внизу). Ці ділянки міцно хімічно прив'язуються до носія. Ми знаємо їх послідовність, ми знаємо з якої хромосоми походить ця ділянка, їх точні координати на склі та назву гену. Це і є чіп.

Біолог виділяє молекули РНК з дослідного зразка та контроля, мітить їх різними флюоресцентними фарбниками (умовно кажучи, червоним і зеленим, малюнок посередині зверху) і наносить на чіп. Я трохи спрощую, щоб не загружати непотрібною інформацією.
Далі відбувається наступне. Ми пам'ятаємо принцип комплементарності зі школи. Дві низки молекул ДНК з'єднуються між собою у подвійну спіраль водневими зв'язками. Якщо нагріти ДНК, вона денатурується і розплітається, якщо трохи охолодити - то з'єднується знову. Під час гібридизації кожна мічена (зеленим або червоним) молекула РНК "шукає" свою відповідну комплементарну ділянку, що нанесена на чіпові і приєднується до неї. Ось тут гарна і проста анімація, де на поверхні стирчить прив'язана ДНК, а до неї чіпляється РНК (насправді її перед цим переписують в кДНК, але це не суть важливо). Потім це все сканують спеціальним сканером і ми бачимо картинку з цятками, які світяться зеленим (якщо цей ген включено в дослідному зразку), червоним (якщо в контролі) і жовтим (якщо ген включений і там, і там). Або це просто одна фарба, якщо ми досліджуємо одну тканину. При цьому інтенсивність світіння відповідає кількості молекул: чим яскравіше - тим сильніше експресується ген.

На виході після сканування ми отримуємо просту таблицю з координатами гену, його назвою і інтенсивністю його роботи в умовних одиницях, екстрагованих з відсканованого зображення. Потім починається рутина по нормалізації, фільтруванню і статистична обробка так, аби в кінцевому рахунку мати список генів, які активно працюють або навпаки, зовсім виключились в дослідному зразку.
Хто нічого не зрозумів, може спокійно пропускати, оскільки цей метод, хоч і був широко розповсюдженим, відходить в минуле (дивимось графік зверху зліва, натомість все активніше застосовується метод другий - секвенування транскриптому.

Робиться точнісінько так, як прочитка геному. Різниця в тому, що замість молекул ДНК біолог виділяє молекули РНК і переписує в ДНК. Хоч вони і невеликі (близько 2000 бп), їх також кришать в дрібну лапшу і читають короткими шматками.
Далі починається та сама весела історія з асемблінгом. Якщо геном прочитаний, то асемблінг роблять на референтний геном. Просто тупо рахують кількість прочитаних шматків РНК, які підходять до певної генної ділянки. До гену Х "приліпилось" 10 шматків, а до гену У - 1000. Вау, ген У сильно експресується. Результати - така самісінька табличка.
Все стає значно сумнішим, якщо у нас нема референтного геному. Тоді біоінформатики роблять de novo асемблінг і потім біолог ридає ридма, напагаючись це інтерпретувати. Правда, перед цим ридають біоінформатики, якщо біолог накосячив і в пробах була контамінація ДНК. Так чи інакше, de novo асемблінг транскриптому має ті самі вади, що асемблінг геному, тільки ще гірше. Не хочу вдаватись в деталі.
Незалежно від того, який метод ми використовували для оцінки експресії генів, у нас на виході така сама табличка, в якій в першій шпальті назва гену, а в наступних - рівень його експресії і статистичні обрахунки.
Ну, і що тепер з ним робити? Далі йде сортування і так, і сяк. Якщо це у нас спостереження процесу в часі, то найчастіше проводять ієрархічну кластеризацію, коли робота генів сортується по профілям. На картинці справа в кольоровій скалі порівняння, скажімо, роботи генів в ракових пухлинах і нормальній тканині. Бачимо групу генів, яка включена там і виключена деінде.

Програм для кластеризації є тьма. Наприклад, тут.
Трохи просунутіші намагаються вибратись з двомірного простору і подивитись на нетворки.

Я особисто не зовсім розумію, як їх інтерпретувати, тому базуюсь переважно на сортуванні генів по біологічним процесам. Найулюбленіший тул для візуалізації експресії рослинних генів Mapman. Тут гени вже сортовані по певним клітинним процесам, а туд дозволяє візуалізувати їх роботу в певному зразку.

Тепер увага. Результати подібних експериментів також складаються в відкриті бази даних. Оскільки кожен ген, якщо він відомий і має унікальний ID, то всі експерименти на нього залінковані незалежно від того, хто цей експеримент робив. Можна прослідкувати роботу гену у всіх відомих опублікованих роботах. Приміром, берем ген Х у кукурудзи (ну я ботанік, тому кукурудза) і можем дивитись його роботу в експериментах з посухостійкості з лаби 1, в експериментах на мутантах з лаби 2, в експериментах по враженню мучнистою росою з лаби 3. Найпотужніший ресурс для цього Genevestigator.
Насправді з усих відкритих даних осмислено можливо 1%. Як це виглядає на практиці. В лабораторії, що спеціалізується на якомусь просцесі робиться цей гігантський скринінг роботи генів якогось досліду. Потім береться один процес і уважніше описується. Решта до уваги не береться не тому, що не хочуть, а тому, що нереально охопити. Але всі дані складаються. Кому цікаво, може брати і колупати далі.
Є більш симпатичніші іграшки, які до того ж відкриті. Тут можна взагалі пропасти. Це візуалізація роботи генів у різних організмів з картинками для розкрасок.

Тепер. Ми. Берем. Геномні і транскриптомні дані, комбінуєм їх і пробуєм з цим всим злетіти.

Це картинка зі статті про прочитку пшеничного геному. Оскільки там три пари геномів і у кожного свої гени, то дослідили їх роботу одномоментно. Виявилось, що ці три пари геномів ще поштовхались в клітині, перш ніж узгодили свою роботу. Кожна пара може існувати сама по собі так, щоб був нормальний організм. А тепер уявіть собі три пари програм в одному організмі. Цей процес називається переплавкою геномів. Один геном попросили заткнутись і не працювати, інший навпаки, активізувався і давай рубати транскрипти запасних білків, третій пробує ці два примирити.
Я сказала білки? Так ось, наступна тема розмови - протеоміка.

{kind=link}
Комментарии (0)